臨床試験情報
JACCRO CC-06(研究代表者:市川 度)
| 項目 | 説明 |
|---|---|
| 試験課題名 | 切除不能進行・再発大腸癌におけるEGFR陽性・KRAS遺伝子野生型に対する一次治療ティーエスワン+オキサリプラチン(SOX)+セツキシマブ併用療法の第I/II相試験 |
| 研究の目的 | 切除不能進行・再発大腸癌におけるEpidermal Growth Factor Receptor(EGFR)陽性・KRAS遺伝子野生型に対する一次治療としてティーエスワン+オキサリプラチン+セツキシマブ併用療法を行い、第I相試験では最大耐用量(MTD:Maximum Tolerated Dose)と推奨用量(RD:Recommended Dose)を決定する。第II相試験では、第I相試験で得られた推奨用量による治療を行い有効性と安全性を評価する。 |
| Phase | 第I相/第II相試験 |
| 適格規準 | 1) 組織学的に大腸癌であることが確認された症例。 2) EGFR陽性の治癒切除不能な進行・再発大腸癌の症例。 3) KRAS遺伝子野生型(codon12、13)の症例。 4) RECISTに基づく測定可能病変を有する症例(RECIST Ver.1.1)。 5) 切除不能の原発巣、および切除不能の遠隔転移またはリンパ節転移の場合は、前治療として化学療法を行っていない症例(手術を施行した場合、手術以外の治療を行っていないこと)。再発例の場合は、原発巣または転移巣に対する手術後の初回再発であり、その再発巣に対しては手術療法も含め治療を行っていない症例(術後補助療法施行例は治療終了6か月以上経過して再発した症例)。 6) ECOGのPerformance Status(PS)が0~1の症例。 7) 同意取得時の年齢が20歳以上(ただし第I相試験は20歳以上79歳以下)の症例。 8) 3か月以上の生存が見込まれる症例。 9) 経口摂取可能な症例。 10) 登録前14日以内の主要臓器機能について、以下の規準を満たしている症例。 ① 白血球数:3,000/mm^3以上、12,000/mm^3以下 ② 好中球数:1,500/mm^3以上 ③ 血小板数:10.0×10^4 /mm^3以上 ④ ヘモグロビン:9.0 g/dL以上 ⑤ 血中ビリルビン:施設基準値上限の1.5倍以下 ⑥ AST:施設基準値上限の2.5倍以下(肝転移を有する場合は5倍以下) ⑦ ALT: 施設基準値上限の2.5倍以下(肝転移を有する場合は5倍以下) ⑧ 血清クレアチニン:1.2mg/dL未満 ⑨ クレアチニン・クリアランス:60 mL/min以上 11) 本試験内容について十分な説明を受け、本人の文書による同意が得られている症例。 |
| 除外規準 | 1) 同時性重複がんまたは無病期間が5年以内の異時性重複がんを有する症例(ただし局所治療で治癒が見込める早期がんは除外とはしない)。 2) 症状を有する脳転移症例。 3) 重篤な感染症を有する症例。 4) 間質性肺炎あるいは肺線維症を有する症例。 5) 重篤な心疾患またはその既往歴を有する症例。 6) 機能障害を伴う重度の感覚異常または知覚不全のある症例。 7) 多量の癌性体腔液(胸水、腹水、心嚢水)を有する症例。 8) 重篤な併存疾患(腎不全、肝不全、高血圧など)を有する症例。 9) 原発巣または転移巣に対して放射線治療が施行された症例。 10) 避妊する意思のない男性。または妊婦、授乳婦、妊娠検査陽性の女性または避妊する意思のない女性。 11) 重篤な過敏症の既往を有する症例。 12) オキサリプラチンまたは他の白金を含む薬剤に対し過敏症の既往歴がある症例。 13) これまでにセツキシマブ、オキサリプラチンまたはティーエスワンの投与を行ったことがある症例。 14) フルシトシンを投与中の症例。 15) 試験責任医師または分担医師が本試験への参加を不適当と判断した症例。 |
| Regimen |
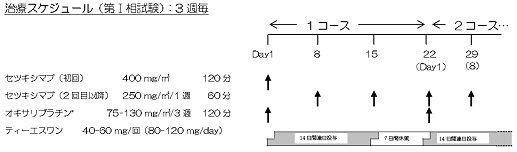
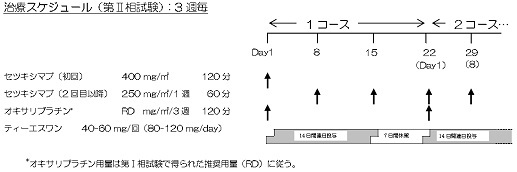
登録後、14日以内にプロトコール治療を開始し、以下の治療法を2週1コースとして「プロトコール治療中止・終了規準」に該当するまで繰り返す。
≪第II相試験≫
|
| 評価項目 |
第I相試験 第II相試験 |
| 目標症例数 | 第I相試験:各レベル 6例、最大12例 第II相試験:56例 第I相試験のRDレベルの症例については、第II相試験に組み込み、その後の追跡調査(最終症例登録後2年間)を実施し、第II相試験の症例として集計する。 |
| 研究期間 | 試験期間:2012年1月~2016年7月(4.5年間) 登録期間:2012年1月~2014年7月(2.5年間) 観察期間:最終症例登録後2年間 |